Technical Deep Dive Agentic Memory (AgeMem) and the Evolution of Unified Memory Management
📎 附件资料
- 在线预览(幻灯片模式):点击打开
- 下载:/assets/agemem_unified_agentic_memory.pdf
🖥️ 幻灯片预览(支持全屏)
1. The Context Window Bottleneck: Reimagining Memory for Long-Horizon Reasoning
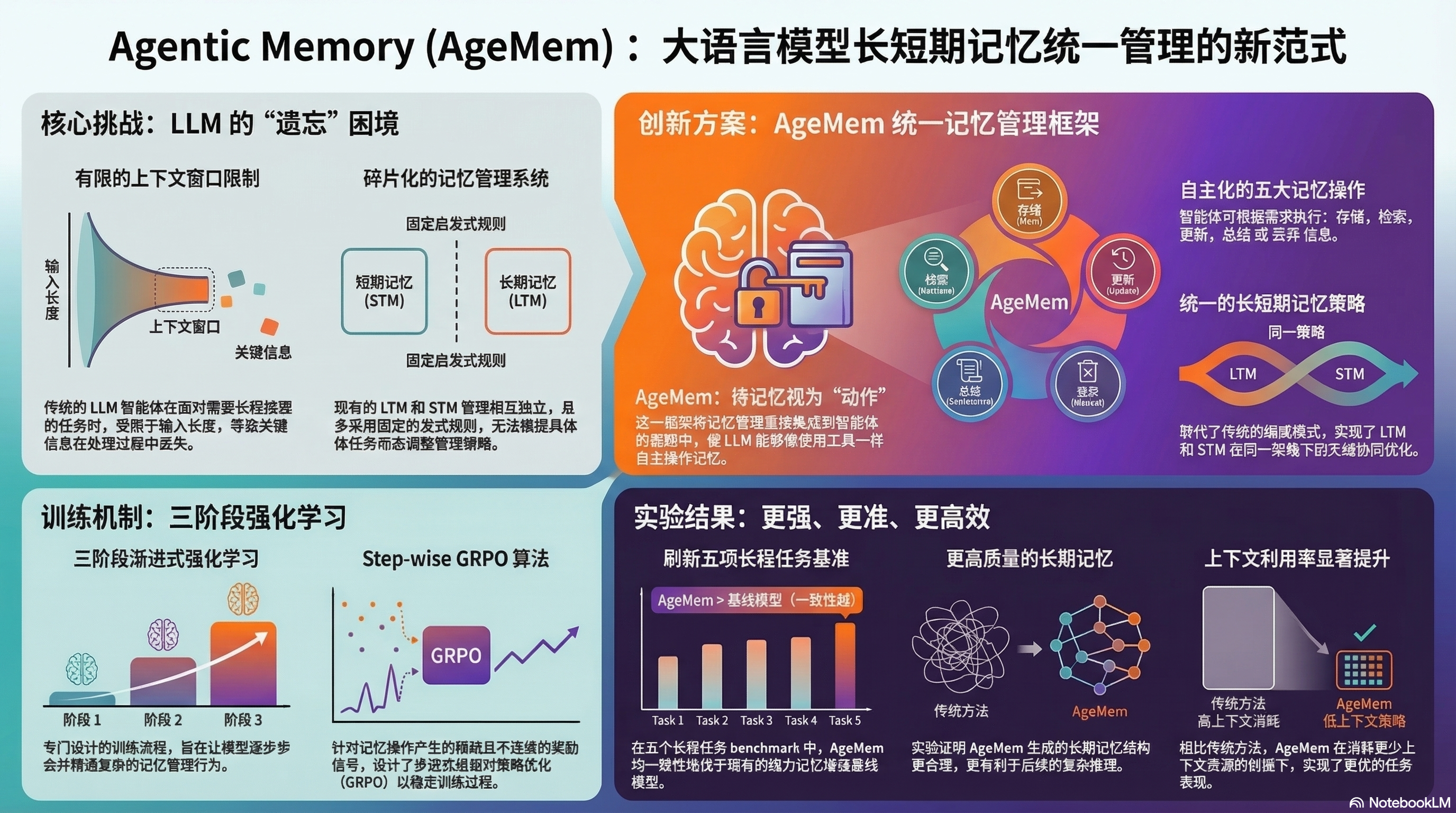
The current landscape of Large Language Models (LLMs) is defined by a persistent architectural constraint: the finite context window. While state-of-the-art models demonstrate remarkable proficiency in short-term information processing, their capacity for long-horizon reasoning—tasks necessitating the maintenance of state over extended temporal scales or complex sub-steps—remains fundamentally tethered to the volume of data that can be actively held in “working memory.” Overcoming these constraints is a strategic imperative for the development of autonomous agents; without sophisticated memory management, agents suffer from rapid performance degradation as critical task context is evicted or diluted.
The research presented by Yu et al. (2026) in “Agentic Memory: Learning Unified Long-Term and Short-Term Memory Management for Large Language Model Agents” addresses a core deficiency in existing systems: the reliance on passive heuristics or external auxiliary controllers. These modular approaches often fail to mitigate state-space fragmentation because they are decoupled from the agent’s primary reasoning policy. Such systems cannot be optimized end-to-end, leading to a “semantic disconnect” where the retrieval mechanism is unaware of the agent’s immediate reasoning requirements. The Agentic Memory (AgeMem) framework resolves this by treating memory management as a first-class citizen within the agent’s decision-making architecture.
2. Architectural Paradigm Shift: From Heuristics to Unified Agentic Policy
AgeMem marks a transition from modular, rule-based memory augmentation to a unified, policy-driven architecture. In this paradigm, Long-Term Memory (LTM) and Short-Term Memory (STM) are not managed by fixed algorithms (like RAG or sliding windows) but are integrated directly into the agent’s action space. This allows the model to harmonize policy gradients across both reasoning and memory-management tokens, ensuring that every internal state transition serves the final objective.
| Feature | Traditional Memory Augmentation | AgeMem Unified Framework | Latency/Overhead |
|---|---|---|---|
| Optimization Method | Modular/Heuristic-based | End-to-end Policy Optimization | Higher (Inference-time logic) |
| Control Logic | Fixed rules or auxiliary controllers | Autonomous, policy-led decision making | Moderate (Action sampling) |
| Adaptability | Rigid; relies on predefined triggers | High; model-agnostic and state-dependent | Low (Native LLM policy) |
| Integration | LTM and STM are separate silos | Unified management within the action space | Minimal (Integrated embedding) |
The critical “So What?” of this architecture lies in its ability to enable end-to-end optimization. By embedding memory management into the agent’s policy, AgeMem allows the LLM to learn the latent utility of its own knowledge base. This eliminates the friction between “reasoning” and “remembering,” as the model can now weigh the cost of memory operations against the probability of task success. This integration facilitates a level of dynamic context steering that was previously impossible under rigid, heuristic-driven regimes.
By conceptualizing memory as an active toolset, the architecture transforms the agent from a passive consumer of context into an active curator of its own cognitive state.
3. Memory as a Toolset: The Taxonomy of Agentic Operations
The AgeMem framework innovates by exposing memory management as a discrete set of tool-based actions. This expansion of the action space allows the agent to exercise granular control over its internal knowledge state. The taxonomy consists of five core operations:
- Store: The agent proactively identifies high-utility information within the current active context and commits it to LTM, ensuring that critical data is not lost when the context window shifts.
- Retrieve: When the current STM is insufficient for the task at hand, the agent triggers a retrieval action to ingest specific, relevant historical data from the LTM back into the active window.
- Update: To mitigate the risk of stale information, the agent can modify existing LTM entries. This ensures state accuracy over time, allowing the agent to correct previous assumptions as new data emerges.
- Summarize: This operation manages state density by compressing high-cardinality information into concise representations, preserving the “semantic essence” while optimizing the token economy.
- Discard: Essential for improving the Signal-to-Noise Ratio (SNR), the agent performs autonomous pruning of irrelevant or redundant data to prevent the cognitive clutter that often degrades long-context performance.
These operations allow the agent to govern its internal knowledge state with high precision. By distinguishing between “Update” (maintaining accuracy) and “Summarize” (managing context density), the agent optimizes its context window for maximum reasoning utility. This autonomous governance ensures that the most pertinent information is always prioritized, effectively extending the functional context window beyond its physical limitations.
Mastering this expanded action space, however, requires a specialized training methodology to overcome the challenges of non-differentiable memory operations.
4. Advanced Training Methodology: Progressive RL and Step-wise GRPO
Training an agent to master discrete memory operations is non-trivial due to the presence of sparse and discontinuous rewards. A “Store” action taken at an early timestep may not yield a discernible reward until much later in the episode, creating a significant credit assignment problem.
To resolve this, Yu et al. (2026) propose a Three-Stage Progressive Reinforcement Learning (RL) Strategy:
- Stage 1: Foundational Skill Acquisition: The agent is trained on supervised memory trajectories (behavioral cloning) to learn the basic mechanics and syntax of the five memory tools.
- Stage 2: Contextual Integration: The agent practices these operations within simplified reasoning environments, learning to trigger specific tools based on the current state-space.
- Stage 3: Unified Policy Refinement: The model undergoes full RL to optimize the interplay between memory operations and final task performance, harmonizing the policy for complex environments.
A pivotal innovation in this stage is Step-wise Group Robust Policy Optimization (GRPO). While standard GRPO (popularized for its efficiency in models like DeepSeek-R1) provides stability without a critic model, the “Step-wise” modification is critical for memory-heavy tasks. It enables intra-episode reward credit assignment, allowing the model to receive granular feedback on memory actions rather than relying solely on a single terminal reward. This prevents policy collapse and ensures that the model learns the delayed utility of specific “Store” or “Retrieve” actions, resulting in a more robust and stable gradient during the optimization process.
The successful implementation of this training regimen leads to a model capable of superior performance across diverse architectural backbones.
5. Empirical Validation: Performance, Quality, and Efficiency Gains
The AgeMem framework was evaluated across five rigorous long-horizon benchmarks. The results demonstrate that giving an agent autonomous control over its memory provides three primary value drivers:
- Task Performance: AgeMem consistently outperformed strong baselines, including traditional RAG and fixed-rule memory systems. On long-horizon reasoning tasks, the model showed a significant delta in performance metrics like Pass@k, proving that agentic control over context is superior to static retrieval.
- Memory Quality: Agentic control yields higher-quality long-term retention. By selectively “Storing” and “Updating” information based on task relevance, the LTM maintained a high SNR, avoiding the information dilution typical of automated heuristic storage.
- Context Efficiency: The framework optimized the limited context window through autonomous pruning and summarization. This improved the token economy, allowing the model to maintain higher ROUGE scores for relevant context while using fewer total tokens.
The empirical data across multiple LLM backbones confirms that AgeMem is model-agnostic, providing a scalable solution for any agentic architecture requiring long-term state maintenance. These findings suggest that the framework’s ability to selectively manage its own knowledge base is a fundamental requirement for the next generation of autonomous AI.
6. Conclusion: The Strategic Implications of Autonomous Memory
The AgeMem framework, as detailed by Yu et al. (2026), redefines the role of memory in AI systems. By shifting from “memory as a static storage” to “memory as an agentic tool,” this research mitigates the fundamental constraints of finite context windows that have long hindered LLM development.
The strategic implication for systems architects is clear: the path to truly autonomous, persistent agents lies in the integration of memory management directly into the core reasoning policy. AgeMem provides the necessary blueprint for this evolution, demonstrating that when an agent is empowered to curate its own knowledge state, it achieves a level of reasoning depth and operational efficiency previously unattainable. This research sets a new standard for agentic architectures, paving the way for AI systems capable of handling the most complex, multi-step workflows in modern computing.